

10.1038/s41596-018-0089-3
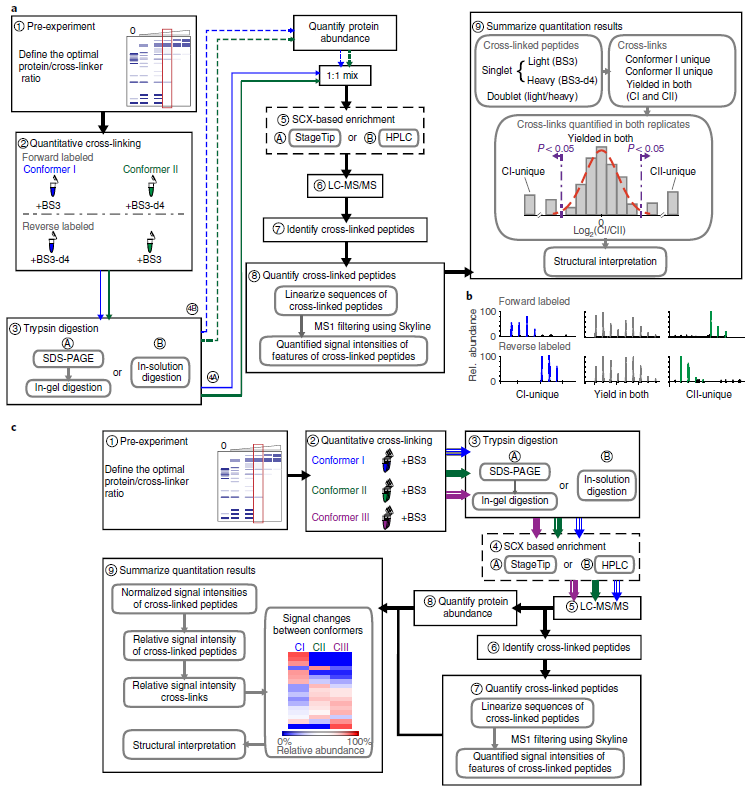
Quantitative cross-linking/mass spectrometry (QCLMS/QXL-MS) probes structural changes of proteins in solution. This method has revealed induced conformational changes, composition shifts in conformational ensembles and changes in protein interactions. It illuminates different structural states of proteins or protein complexes by comparing which residue pairs can be cross-linked in these states. Cross-links provide information about structural changes that may be inaccessible by alternative technologies. Small local conformational changes affect relative abundances of nearby cross-links, whereas larger conformational changes cause new cross-links to be formed. Distinguishing between minor and major changes requires a robust analysis based on carefully selected replicates and, when using isotope-labeled cross-linkers, replicated analysis with a permutated isotope-labeling scheme. A label-free workflow allows for application of a wide range of cross-linking chemistries and enables parallel comparison of multiple conformations. In this protocol, we demonstrate both label-free and isotope-labeled cross-linker-based workflows using the cross-linker bis(sulfosuccinimidyl)suberate (BS3). The software XiSearch, developed by our group, is used to identify cross-linked residue pairs, although the workflow is not limited to this search software. The open-access software Skyline is used for automated quantitation. Note that additional manual correction greatly enhances quantitation accuracy. The protocol has been applied to purified multi-protein complexes but is not necessarily limited to that level of sample complexity. Optimizing the cross-linker/protein ratio and fractionating peptides increase the data density of quantified cross-links, and thus the resolution of QCLMS. The entire procedure takes ~1-3 weeks.