

https://doi.org/10.1021/acs.jproteome.8b00600
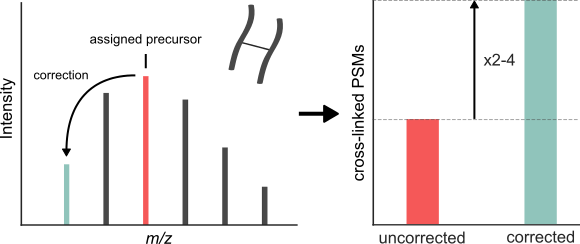
Cross-linking/mass spectrometry has undergone a maturation process akin to standard proteomics by adapting key methods such as false discovery rate control and quantification. A poorly evaluated search setting in proteomics is the consideration of multiple (lighter) alternative values for the monoisotopic precursor mass to compensate for possible misassignments of the monoisotopic peak. Here, we show that monoisotopic peak assignment is a major weakness of current data handling approaches in cross-linking. Cross-linked peptides often have high precursor masses, which reduces the presence of the monoisotopic peak in the isotope envelope. Paired with generally low peak intensity, this generates a challenge that may not be completely solvable by precursor mass assignment routines. We therefore took an alternative route by ‘”in-search assignment of the monoisotopic peak” in the cross-link database search tool Xi (Xi-MPA), which considers multiple precursor masses during database search. We compare and evaluate the performance of established preprocessing workflows that partly correct the monoisotopic peak and Xi-MPA on three publicly available data sets. Xi-MPA always delivered the highest number of identifications with ∼2 to 4-fold increase of PSMs without compromising identification accuracy as determined by FDR estimation and comparison to crystallographic models.